Autores
- Silva, Ana I. (1); Almirón, Florencia (1); Ferreira, Elizabeth (1); Pistón, Mariela (2)
- Departamento de Metrología Química, Laboratorio Tecnológico del Uruguay, Montevideo, Uruguay.
- Grupo de Análisis de Elementos Traza y Desarrollo de Estrategias Simples para Preparación de Muestras (GATPREM), Área Química Analítica, Departamento Estrella Campos, Facultad de Química, Universidad de la República, Montevideo, Uruguay
Los métodos primarios constituyen el nivel jerárquico de medición más elevado debido a que permiten implementar la trazabilidad a las unidades del Sistema Internacional. El objetivo de este trabajo fue la optimización de un método primario para la determinación de creatinina en suero humano por dilución isotópica mediante cromatografía gaseosa acoplada a espectrometría de masa (GC – IDMS), adaptado de un método de referencia publicado en el Joint Committee for Traceability in Laboratory Medicine (JCTLM - BIPM). Se optimizaron factores vinculados a las etapas de equilibrado isotópico y derivatización. Para esto se realizó un diseño experimental de tres factores en dos niveles (diseño 23) y un análisis de varianza de un factor (ANOVA). Las respuestas evaluadas en el diseño experimental fueron la señal correspondiente a la creatinina, la eficiencia de derivatización, la relación de eficiencia de derivatización y el sesgo. El método propuesto, optimizado para las etapas de equilibrado isotópico y derivatización, presenta mayor eficiencia y simplicidad comparado con el método original, y cuenta con un sesgo adecuado al propósito. Luego de su validación podrá ser implementado para el desarrollo de herramientas de aseguramiento de la calidad de los laboratorios clínicos que realizan estas mediciones rutinariamente.
Palabras clave: metrología química, métodos primarios de medición, IDMS optimización.
Introducción
La armonización de las mediciones a nivel global es crucial para el comercio, la salud y el avance tecnológico y científico. La metrología, es decir, la ciencia que estudia las mediciones y su aplicación, trabaja activamente para alcanzar este objetivo. Probablemente, la forma más económica de alcanzar la comparabilidad global de las mediciones sea el desarrollo de un sistema jerárquico de medición basado en la trazabilidad mediante estándares de calibración a una referencia internacional adecuada, como el Sistema Internacional de Unidades (Thompson y Ellison, 2005). Este sistema de medición depende críticamente de incertidumbres bajas entre los niveles, las cuales se alcanzan mediante mediciones de alta jerarquía metrológica. El vocabulario internacional de metrología (VIM) define la trazabilidad metrológica como la “propiedad de un resultado de medida por la cual el resultado puede relacionarse con una referencia mediante una cadena ininterrumpida y documentada de calibraciones, cada una de las cuales contribuye a la incertidumbre de medida” (JCGM, 2012). La trazabilidad metrológica a las unidades del sistema internacional se obtiene a partir de los métodos primarios, que representan el mayor nivel metrológico. Estos cuentan con ecuaciones e incertidumbres descritas completamente utilizando unidades del sistema internacional (King, 2005) y no tienen correcciones significativas por factores empíricos (De Bievre y Peiser, 1997). En el campo de la metrología química, la unidad aplicable es el mol y su trazabilidad se obtiene por métodos primarios como la gravimetría y la dilución isotópica con espectrometría de masas (IDMS), que cuenta con demostrada veracidad y baja incertidumbre. Estas características le confieren especial relevancia para la certificación de materiales de referencia y en la asignación de valores de referencia de ensayos de aptitud. Este método, de vasta aplicación en el ámbito de la metrología química orgánica, consiste en adicionar a la muestra una cantidad conocida del analito de interés que presenta una composición isotópica diferente y que cumple la función de estándar interno. La determinación de la cantidad de sustancia se calcula a partir de una ecuación que depende de la relación entre la abundancia isotópica natural del analito y la abundancia isotópica del estándar interno enriquecido isotópicamente, y esta se pueden medir con alta exactitud mediante un espectrómetro de masas (Richter, 1997). Para que este método alcance su altísimo potencial analítico, es fundamental que parámetros como la preparación de la muestra o la selección del estándar interno sean estudiados y optimizados exhaustivamente.
Un ejemplo de aplicación para este tipo de método primario es la determinación de creatinina en suero por dilución isotópica con espectrometría de masas.
La importancia de la determinación de la concentración de creatinina en suero se debe a que es utilizada clínicamente para la estimación de la tasa de filtración glomerular con el fin de diagnosticar la enfermedad renal crónica. Se considera que esta enfermedad afecta a entre 5 -10 % de la población mundial, con un aumento sostenido de la incidencia y prevalencia de la enfermedad renal en etapa terminal, la falla renal y los trasplantes de riñón a nivel mundial (Myers, et al., 2006). De forma rutinaria, este análisis se realiza por métodos colorimétricos como el método de Jaffe, que se ve afectado por interferencias de otros grupos cromóforos provenientes de la bilirrubina, proteínas y cetonas (Fernández-Fernández, et al., 2014). Estas deficiencias se compensan parcialmente tras el desarrollo de métodos enzimáticos (Myers, et al., 2006).
En el campo de la bioquímica clínica, el diagnóstico, la evaluación de riesgo y el monitoreo de un paciente dependen de forma crítica de los resultados emitidos por un laboratorio (Thienpont, et al., 2002). La comparabilidad de estos resultados permite considerar intervalos de referencia comunes o posibles estrategias de tratamiento (Thienpont, et al., 2002). Esta comparabilidad de resultados se obtiene principalmente al establecer la cadena de trazabilidad de la medición a los estándares internacionales más altos, mediante un sistema de mediciones de referencia exhaustivo (Thienpont, et al., 2002).
El Comité Conjunto para Trazabilidad en Medicina Laboratorial (JCTLM según sus siglas en inglés) lista los métodos de referencia existentes para la determinación de parámetros clínicos, muchos de los cuales emplean dilución isotópica (Kessler, 2016). La misión general de este comité, que forma parte de la Oficina Internacional de Pesas y Medidas (BIPM, según sus siglas en francés) es la mejora de la calidad en el ámbito de la salud, reduciendo los costos del gobierno y la industria del diagnóstico in vitro por medio de la promoción de sistemas de examinación de referencia que permiten la trazabilidad de los resultados en los exámenes y la comparabilidad mejorada. Complementariamente a la implementación de la trazabilidad en los laboratorios clínicos, la participación en instancias de evaluación externa así como la aplicación de herramientas de aseguramiento de calidad internas son elementos clave y mandatorios para estos laboratorios.
Los resultados de programas de ensayos de aptitud permiten evaluar el desempeño individual de cada laboratorio y la comparación de métodos analíticos comúnmente utilizados. La asignación de valor de las muestras distribuidas en estos estudios, usualmente muestras liofilizadas basadas en suero humano, se realiza mediante el empleo de un método de referencia o de un material de referencia certificado. En caso de que esto no sea posible, se puede instrumentar la asignación de valor por consenso, lo que presenta un inconveniente mayor dado que no es posible contar con un valor metrológicamente trazable, por lo tanto no presenta un punto de comparación a nivel internacional. Esto dificulta la determinación certera de los sesgos asociados a los diferentes métodos analíticos.
Actualmente, los valores de referencia de los programas de evaluación externa de calidad ofrecidos en Uruguay son asignados externamente por métodos de referencia secundarios, lo cual resulta en un elevado costo asociado al transporte. El desarrollo o implementación de métodos primarios de trazabilidad claramente establecida permitirá la asignación de valores trazables a estos programas para realizar una evaluación rigurosa de la exactitud de los métodos de rutina utilizados a nivel nacional.
Este trabajo introduce modificaciones a un método de referencia para la determinación de creatinina por suero humano mediante dilución isotópica con cromatografía gaseosa acoplada a espectrometría de masas (GC-IDMS) publicado en la base de datos del Joint Committee for Traceability in Laboratory Medicine (JCTLM) del BIPM (Siekmann, 1985). Las modificaciones propuestas al método son el uso de un estándar interno deuterado (creatinina-d3) en sustitución del original (creatinina-13C,15N2) y la preparación gravimétrica de las muestras y calibrantes.
El estándar interno presentado como alternativa se obtiene fácilmente de forma comercial, a diferencia del original, que debe sintetizarse especialmente y requiere de equipamiento no siempre disponible en un laboratorio analítico.
Adicionalmente, la preparación gravimétrica de muestras y calibrantes le confiere al método mayor exactitud y menor incertidumbre, al tiempo que elimina la necesidad de calibrar el equipamiento volumétrico. Dado que la elección de estándar interno utilizado puede incidir fuertemente en el desempeño de los métodos basados en dilución isotópica, es necesaria la búsqueda de condiciones óptimas para que su veracidad no se vea afectada.
El objetivo de este trabajo es evaluar el efecto de factores relacionados con etapas críticas de preparación de muestra, como el equilibrado isotópico y la derivatización, sobre este método optimizado con los cambios propuestos. A partir de esta información se busca encontrar las condiciones óptimas para estas etapas para que el método presente una veracidad adecuada al propósito de desarrollar herramientas de aseguramiento de calidad.
Materiales y Métodos
Reactivos y materiales
Para la cuantificación de las muestras de suero se utilizó un material de referencia certificado de creatinina pura (SRM 914a, NIST, Maryland, USA), un estándar de creatinina-d3 (sc-217956, Santa Cruz Biotechnology Inc., Texas, USA), y agua ultrapura “agua MQ” producida por un sistema de ultrapurificación DirectQ3-UV Millipore (Merck KGaA, Darmstadt, Alemania).
Para el diseño experimental se utilizaron materiales de referencia certificados de creatinina en suero congelado (SRM 967a, NIST, Maryland, USA). El filtrado de las muestras se realizó con filtros de 0,45 µm.
Se utilizó ácido acético glacial (Macron Fine Chemicals, Pensilvania, USA) para preparar una solución de ácido acético al 1% “SAc” (1 mL de ácido acético se llevó a 100 mL en un matraz aforado con agua MQ) e hidróxido de amonio 30% (Carlo Erba, Milán, Italia) para preparar una solución de hidróxido de amonio 2M “SB” (11,3 mL NH4OH se llevaron a 100 mL en matraz aforado con agua MQ).
Para la separación de creatina y creatinina se utilizó una resina de intercambio catiónico en forma hidrógeno, fuertemente ácida, 200 – 400 mesh DOWEX 50WX2 (Sigma-Aldrich, Misuri, USA), en una columna de vidrio de 12,5 mm de diámetro interno y 15 cm de largo. El control de pH de las fracciones recolectadas se realizó con tiras de pH en el rango 0 a 14 (código 109535) y 2,5 a 4,5 (código 109541) (Merck KGaA, Darmstadt, Alemania). Para el secado de las muestras se utilizó nitrógeno calidad 4.0. Durante la derivatización de las muestras se utilizó piridina (secada con tamiz molecular 0,3 nm código 105734 Merck KGaA, Darmstadt, Alemania) y N-Metil-N-(trimetilsilil) trifluoroacetamida (MSTFA, código 111805, Merck KGaA, Darmstadt, Alemania) o N, O-Bistrifluoroacetamida (BSTFA) con 1% Trimetilclorosilano (TMCS) (código 15238 Fluka, Misuri, USA), según corresponda (ver sección “Optimización de solvente y agente de derivatización” en Resultados y discusión).
Equipamiento
Las muestras y soluciones calibrantes se pesaron en una microbalanza Mettler Toledo XPE56 (Zúrich, Suiza). Las muestras de suero humano se manipularon dentro de una cabina de bioseguridad (BSC-1300 II A2-X, Labotecgroup.com) y se llevaron a sequedad con un evaporador de muestras de bloque seco (código 109A 28680-30, Cole-Parmer, USA). La derivatización se realizó en una estufa (modelo FD 115, Binder, Alemania). Las muestras fueron analizadas en un cromatógrafo de gases 7890A acoplado a espectrómetro de masa 5795C (Agilent technologies, USA). El análisis fue realizado en las siguientes condiciones: columna de 5% fenilmetilpolisiloxano de 30 m x 0,25 mm x 0,25 µm (HP-5 MS, Agilent technologies, USA). Se utilizó helio como gas portador a un flujo de 1 mL/min. La temperatura inicial de la columna fue 80 °C por 1 minuto, luego aumentó a 10 °C/min hasta 200 °C y 25 °C/min hasta 250 °C por 5 minutos. La inyección de 0,5 μL se realizó en un modo splitless a 300 °C. La temperatura de la interfase y la fuente de iones del espectrómetro de masa fueron 250 °C y 230 °C, respectivamente. La cuantificación de creatinina se realizó en modo “Single Ion Monitoring”, monitoreando los iones 329 m/z y 332 m/z. Para el análisis de eficiencia de derivatización, también se monitorearon los iones 256 m/z y 259 m/z. La determinación de humedad en piridina fue realizada utilizando un titulado culombimétrico Karl – Fisher C20 (Mettler Toledo, Zúrich, Suiza).
Procedimiento
Se prepararon gravimétricamente soluciones calibrantes a partir de 3 mg de material de referencia certificado de creatinina con 25 g de agua MQ, a razón de dos soluciones independientes por cada instancia de cuantificación. Estas soluciones se diluyeron gravimétricamente para preparar soluciones de trabajo de 5; 7; 8,5; 9 y 11 mg kg-1. La solución de creatinina isotópicamente marcada (creatinina-d3, de ahora en adelante se llamará solución de isótopo) se preparó de forma equivalente a las soluciones calibrantes pero partiendo de 1,5 mg del sólido. Se tomó 1 mL de suero humano previamente descongelado y homogeneizado o 1 mL de solución calibrante y se les agregó gravimétricamente solución de isótopo de manera que iguale la masa de creatinina presente en las muestras analizadas y la solución calibrante a 8,5 mg kg-1.
En adelante y para simplificar, las mezclas de solución calibrante y solución isótopo se denominarán BZY y las mezclas de suero y solución calibrante BXY. Para alcanzar el equilibrio isotópico, estas muestras fueron homogeneizadas por un tiempo variable entre 30 y 90 min (factor “tI”) en un agitador orbital a temperatura ambiente y protegidas de la luz. Las soluciones calibrantes se reservaron para posterior secado, mientras que las muestras de suero humano se trasvasaron a una jeringa sin aguja y se filtraron a 0,45 µm hacia un tubo pyrex de 50 mL. Se agregaron 20 a 25 gotas de SAc a cada muestra, hasta comprobar un pH ~ 4,5.
Para el intercambio iónico se acondicionó una cucharada de resina en un matraz Erlenmeyer con 5 mL de SB, verificando pH básico. Se prepararon las columnas cromatográficas adicionando 2,5 mL de resina y realizando tres lavados de 10 mL de agua MQ hasta comprobar pH neutro. Se sembraron las muestras y se procedió a lavar la creatina con dos tomas de 10 mL de agua MQ. Se recolectó la creatinina adicionando 10 mL de SB, descartando los primeros 2 mL y recolectando la fracción de creatinina en los próximos 8 mL. Las fracciones de creatinina y las soluciones calibrantes se secaron en evaporador bajo nitrógeno utilizando un bloque de calor seco a 80 °C. Una vez que las muestras y soluciones calibrantes se encontraron completamente secas, se les agregó 200 μL de piridina y 50 μL de MSTFA o BSTFA + 1% TMCS, según corresponda (ver sección “Optimización de solvente y agente de derivatización” en Resultados y discusión). Se homogeneizaron mediante vórtex por 10 segundos y se trasvasaron utilizando una pipeta Pasteur de vidrio a un inserto de 400 μL. Luego, estos se colocaron en viales de automuestreador de 2 mL y se calentaron en estufa por un tiempo variable de entre 20 y 70 minutos (factor “tD”) a una temperatura variable de entre 40 y 80 °C (factor “TD”). Posteriormente, las muestras se analizaron por GC-MS.
Optimización de solvente y agente de derivatización
Tres tomas de piridina fueron sometidas a diferentes condiciones de secado. En el primer caso se adicionó tamiz molecular 72 horas antes de su uso (“PIR 72h”), en el segundo caso 4 horas antes (“PIR 4h”) y en el tercer caso no se adicionó tamiz (“PIR ST”). Cuando aplicó, se adicionó tamiz en una proporción 1 mL de tamiz seco por cada 25 mL de solvente. Se prepararon 15 mL de una mezcla BXY de 20 mg kg-1de creatinina y creatinina-d3 y se realizaron tomas de 1 mL en 12 viales de 4 mL ámbar, para analizar cada variación por triplicado.
Las muestras se secaron según lo descrito en la sección anterior y se les agregó 200 μL de piridina (PIR 72h, PIR 4h o PIR ST, según el caso) y 50 μL de MSTFA o BSTFA + 1% TMCS, según corresponda (ver sección “Optimización de solvente y agente de derivatización” en Resultados y discusión). De forma paralela, se determinó la humedad de las muestras de piridina que recibieron diferentes tratamientos de secado por titulación culombimétrica Karl-Fisher por quintuplicado.
Se registró la señal correspondiente al área del ion 329 m/z y se contrastó con el contenido de agua en cada muestra de piridina a través de un análisis de varianza de un factor (ANOVA, nivel de significancia = 5%) mientras que el efecto de la selección de agente derivatizante en la señal creatinina se evaluó mediante un test t de Student (dos colas, nivel de significancia = 5%).
Optimización de tiempo de equilibrado isotópico, tiempo y temperatura de derivatización
La optimización de las variables tiempo de equilibrado isotópico (“tI”), tiempo y temperatura de derivatización (“td” y “TD”, respectivamente) se realizó a través de un diseño factorial de tres factores a dos niveles (diseño 23). Los experimentos realizados se detallan en la Tabla 1.
Tabla 1. Diseño experimental y codificación para cada experimento (entre paréntesis). tI: tiempo de equilibrado isotópico, td: tiempo de derivatización (min) y TD: temperatura de derivatización (°C).
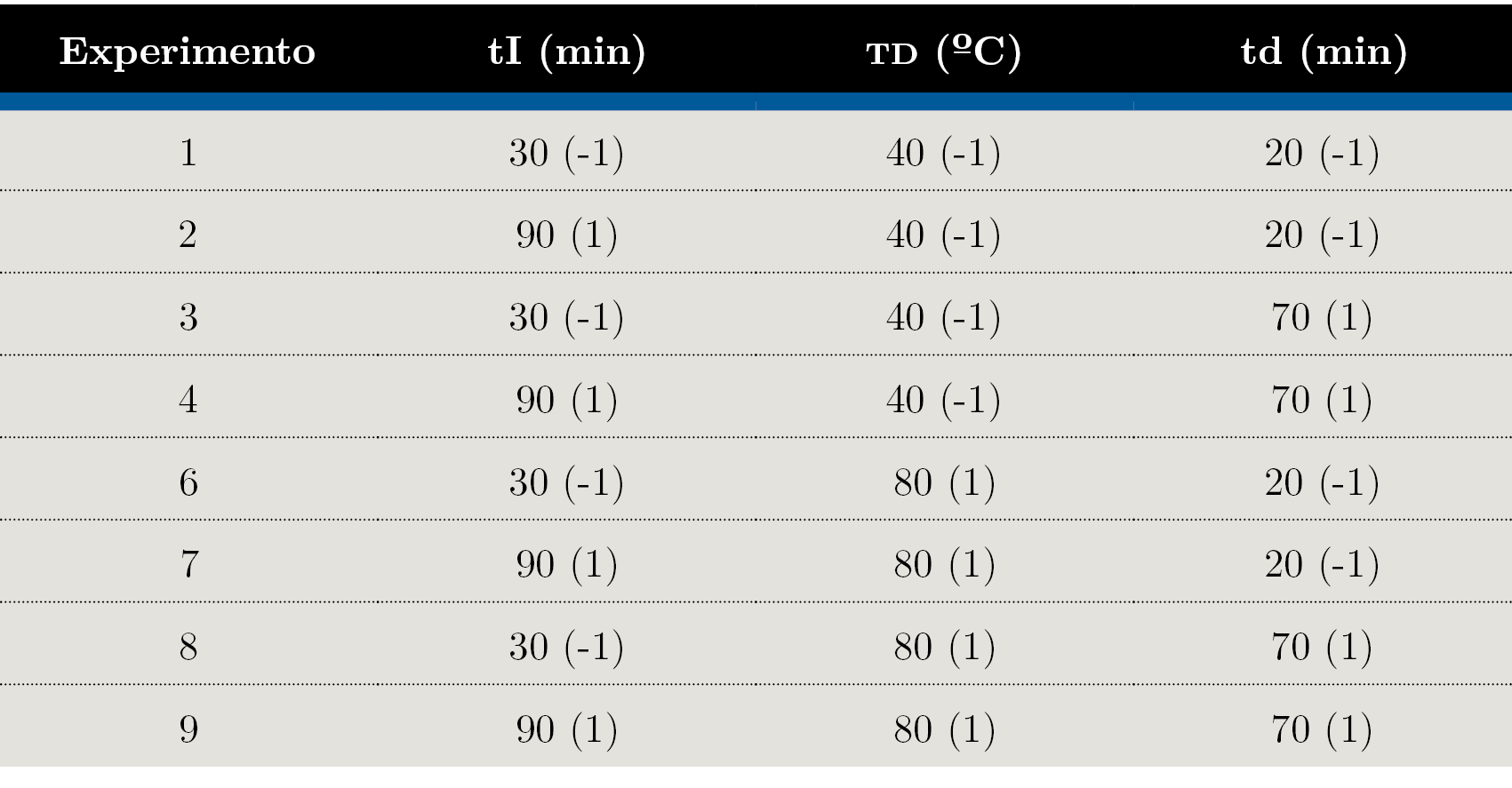
Para llevar a cabo el diseño experimental se analizaron ocho tomas de 1 mL del material de referencia certificado de creatinina en suero humano y se procesaron según el procedimiento detallado anteriormente, variando en cada caso los factores. Se utilizaron materiales de referencia certificados con el objetivo de detectar aquellos experimentos donde el sesgo del método fue acorde al propósito. Las respuestas evaluadas fueron la señal relacionada a creatinina (área de ion 329 m/z), el sesgo determinado como la diferencia entre la fracción de masa certificada y la fracción de masa resultante, la eficiencia de derivatización y la relación de eficiencias de derivatización (“relación ED” definido como el ratio 329/256 m/z: 332/259 m/z). La fracción de masa de creatinina fue calculada por double exact-matching isotope dilution, según la Ecuación 1 (Burke y Mackay, 2008).
$$w_x =w_z\frac{{m_Ym_Z}{(R_Y-R_B)(R_{B,c}-R_Z)R_X}}{m_Xm_{Y,c}(R_B-R_X)(R_Y-R_{B,c})R_Z}$$ (1)
donde wX es la fracción de masa de creatinina en la muestra de suero humano, wZ es la fracción de masa de creatinina en la solución calibrante, mY es la masa de solución de creatinina-d3 adicionada a la mezcla de muestra (BXY), mY,c es la masa de solución de creatinina-d3 adicionada a la mezcla de calibración (BZY), mZ es la masa de solución de calibrante adicionada a la mezcla de calibración (BZY), mX es la masa de suero tomada, R son los ratios de los iones 329 m/z sobre 332 m/z en BZY (RB), en BXY (RB,c), en la solución calibrante (RX y RZ) y la solución del isótopo (RY).
La eficiencia de derivatización fue evaluada como la relación entre la señal del producto tri-derivatizado (tres unidades del grupo trimetrilsilil por molécula de creatinina, “3-TMS-creatinina”) y el producto di-derivatizado, “2-TMS-creatinina”. Esto corresponde a las señales de los iones 329 m/z y 256 m/z, respectivamente. La eficiencia de derivatización también se estudió para el estándar interno, creatinina-d3, en este caso monitoreando los iones 332 m/z y 259 m/z.
El análisis del efecto de los factores individuales y combinados se realizó según las Ecuaciones 2 y 3 (Costa Ferreira, 2015).
$$Efecto factor A=\frac{{\Sigma Y_{+A}}-{\Sigma Y_{-A}}}{4}$$ (2)
$$Efecto\:interacci\acute{o}n\:AB=\frac{{\Sigma Y_{+A{^*}B}}-{\Sigma Y_{-A{^*}B}}}{4}$$ (3)
donde y es la variable de respuesta obtenida, e Y+A y Y+A indican aquellas respuestas y obtenidas cuando el factor A recibió una codificación positiva o negativa, respectivamente. El denominador indica los pares de respuestas Y+A ,Y-A presentes en este diseño de ocho experimentos. Para el estudio de la interacción entre los factores A y B se toman las respuestas Y+A*B ,Y-A*B obtenidas cuando el factor de la codificación de A y B fue positivo o negativo, respectivamente. Numéricamente, cuanto mayores sean estos resultados, mayor es el efecto del factor o interacción de los factores en el proceso estudiado. A su vez, cuando un efecto es positivo, la respuesta aumenta a medida que aumenta el factor y viceversa. La elección de las condiciones óptimas se realizó con un sistema de puntuación diseñado especialmente mediante el cual se asignó un punto por cada condición de optimización cumplida en el experimento, estudiada para cada variable de respuesta. Luego estos puntos se sumaron. Las condiciones de optimización por variable de respuesta fueron seleccionadas como aquellos factores o combinación de factores que presentaron un efecto significativo sobre cada variable de respuesta.
Resultados y Discusión
Optimización de solvente y agente de derivatización
Con el objetivo de compatibilizar la determinación de creatinina con la cromatografía gaseosa como método separativo, fue necesario favorecer la volatilización de este analito mediante derivatización. La optimización de esta etapa impacta directamente en la sensibilidad del método, dado que es posible obtener una mayor señal a partir de la misma cantidad de analito. Las condiciones óptimas fueron analizadas variando la cantidad de humedad presente en el solvente (piridina) y el agente derivatizante utilizado. La humedad es un parámetro crítico a optimizar dado que los productos de derivatización alquil-sililados son sensibles a la humedad y afectan directamente el rendimiento de la reacción (Cardinael, et al., 2015).
Los resultados del estudio de optimización se muestran en la Tabla 2.
Tabla 2. Resultados del estudio de optimización de la etapa de derivatización. PIR 72h, PIR 4h y PIR ST corresponde a los tratamientos de secado aplicado al solvente piridina, descritos en la sección Optimización de solvente y agente de derivatización dentro de Materiales y Métodos.
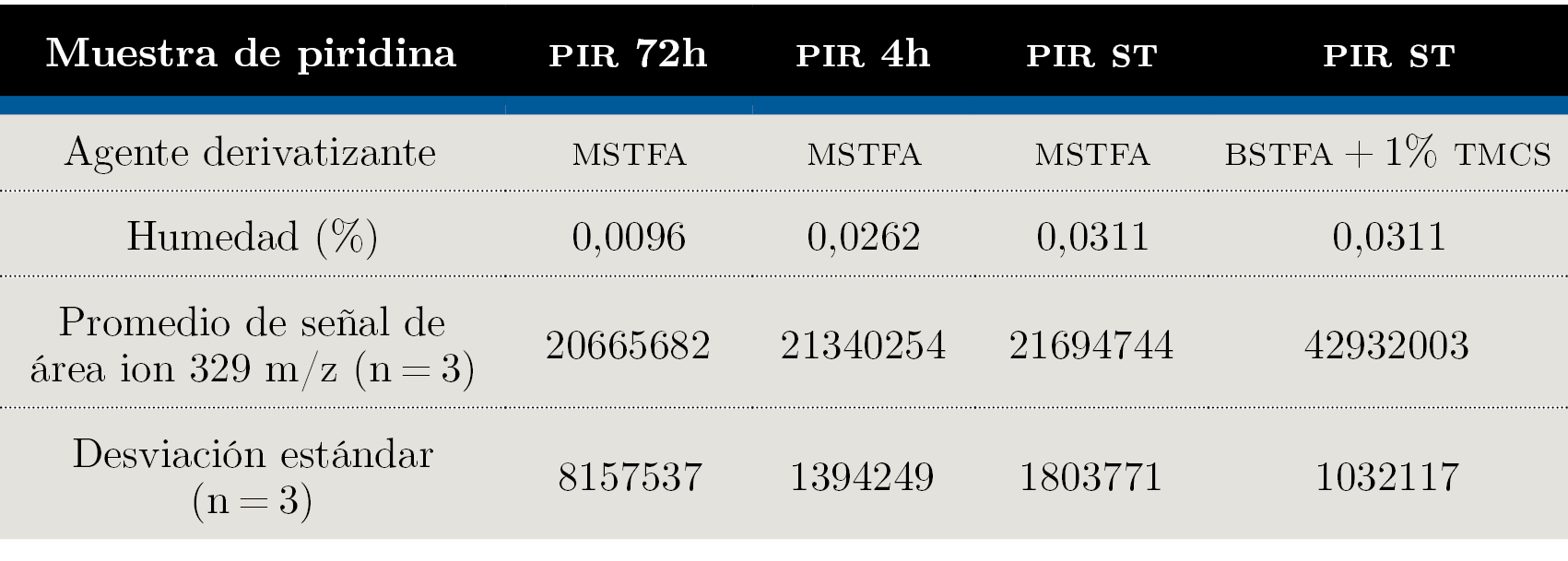
El test de ANOVA indica que la variación de la señal correspondiente a creatinina no puede explicarse estadísticamente por la variación de la humedad del solvente (F< F crítico, α = 5%) evaluado hasta una humedad del 0,04%. Por otro lado, la señal de creatinina se duplicó al utilizar BSTFA + 1% TMCS en contraste con MSTFA, lo que evidencia el efecto de la selección del agente de derivatización sobre la señal de creatinina (ANOVA, F> F crítico, α = 5%). Esto es esperable ya que el agente BSTFA presenta una reactividad mayor que el MSTFA como donador de grupo silil, mientras que la adición de trimetilclorosilano (TMCS) en pequeñas proporciones acelera la reacción con grupos retadores como amidas, aminas secundarias y grupos hidroxilos presentando impedimento estérico (Poole, 2013).
Optimización de tiempo de equilibrado isotópico, tiempo y temperatura de derivatización
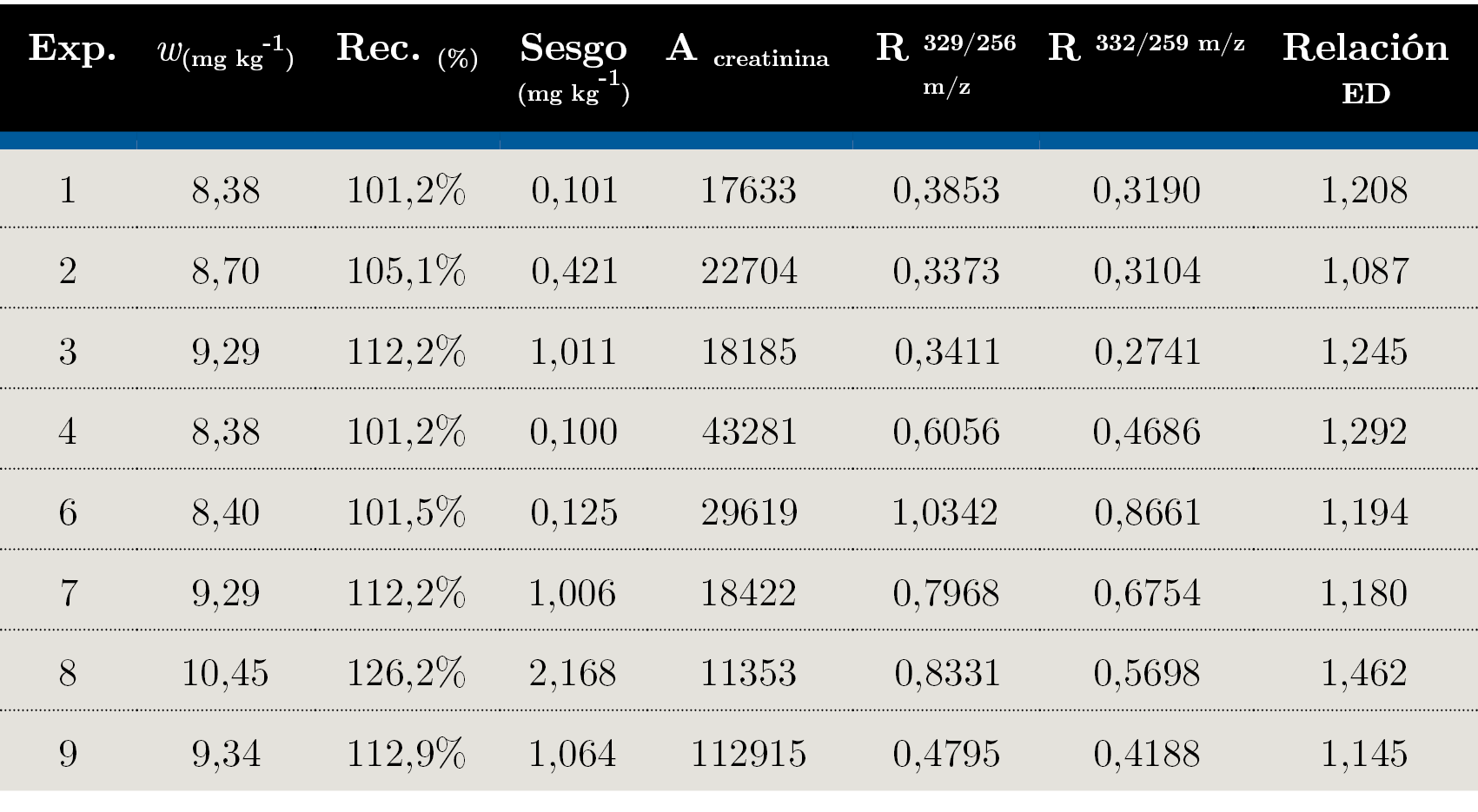
Las variables de respuesta, área de creatinina y eficiencia de derivatización fueron seleccionadas dado que afectan directamente la sensibilidad del método. La variable sesgo evalúa la veracidad del método y su signo toma especial relevancia porque la creatina, presente naturalmente en el suero humano, representa una interferencia del método debido a que tiene el mismo producto de derivatización que la creatinina. La correcta separación entre creatinina y creatina durante la etapa de intercambio catiónico fue previamente estudiada. Adicionalmente, es importante optimizar los factores para buscar que la relación de eficiencias de derivatización entre analito y estándar interno se aproxime a la unidad. El apartamiento de esta condición es un indicio de que el analito y el estándar interno no presentan comportamientos químicos equivalentes, requisito de esencial importancia para la correcta aplicación de la técnica de dilución isotópica. Los resultados obtenidos en el diseño factorial se detallan en la Tabla 3.
Tabla 3. Fracción de masa de creatinina determinada por double exact-matching isotope dilution (Ecuación 1), porcentaje de recuperación que toma como referencia el valor certificado del MRC SRM 967a y cálculo del sesgo, la diferencia entre concentración certificada y fracción de masa calculada.
El efecto de los factores tI, td y TD sobre las variables de respuesta área de creatinina, sesgo, eficiencia de derivatización y relación de ED se calcularon según las Ecuaciones 2 y 3 y se detallan en la Tabla 4.
Tabla 4. Evaluación del efecto de los factores individuales y combinados tI, TD y td sobre las variables de respuesta área de creatinina, sesgo, eficiencia de derivatización y relación ED.
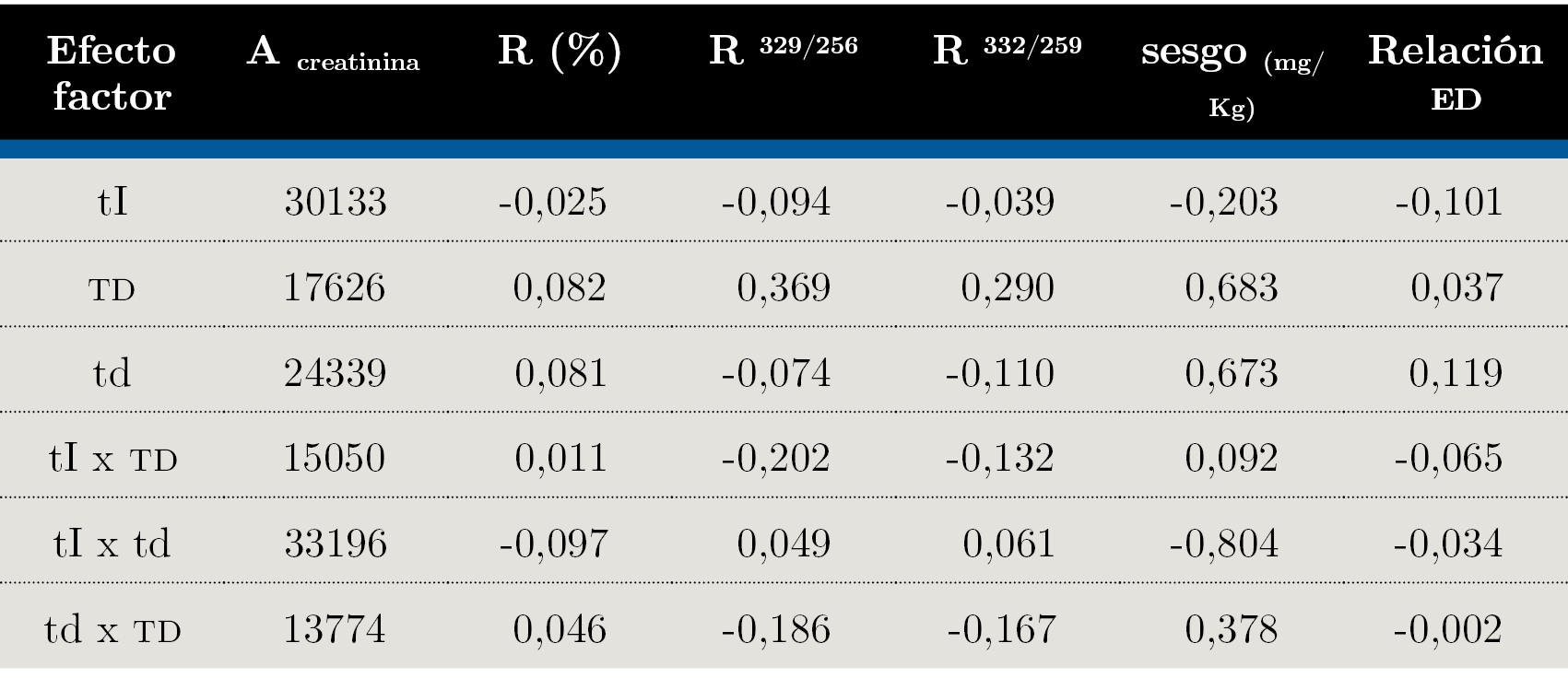
Los tres factores, tI, TD y td, tuvieron un efecto positivo sobre la señal de área de creatinina, tI, en mayor medida que el resto (experimentos 2, 4, 7 y 9: “E2”, “E4”, “E7” y “E9”). Es posible que este aumento se deba a una mejora en la sensibilidad del método o a la presencia de una interferencia como podría ser el caso de E9, con una recuperación de 112%. Además, el efecto combinado de tI y td tiene un mayor impacto en el área de creatinina. Los experimentos donde esto ocurre son E1, E4, E6 y E9. La incidencia de tiempo de derivatización en el aumento de la señal de creatinina puede explicarse por un aumento en la eficiencia de derivatización, produciendo una mayor proporción de 3-TMS-creatinina y aumentando su respectiva señal.
Los resultados obtenidos presentaron sesgos variables, en algunos casos aceptables (recuperación = 101%, E1, E4 y E6) o en otros, sobrecuantificaciones mayores (recuperación = 126%, E8). Dado que en todos los casos se obtuvo un sesgo positivo y recuperaciones mayores a 100%, es importante evaluar qué factor genera un efecto negativo en sesgo y recuperación. Este es el caso de tI y el efecto combinado de tI y td en mayor medida, mientras que el aumento de td y TD afectó negativamente el sesgo. Los experimentos donde el tI presenta un nivel +1 y el producto de tI x td es positivo son E1, E4 y E6. Mediante el equilibrado isotópico se busca que el analito y el estándar interno (creatinina-d3) alcancen la equivalencia química en cuanto a su disposición en la matriz. Si esto no es alcanzado, es posible que el estándar interno se encuentre disponible en menor o mayor grado que el analito, generando una extracción diferencial de los mismos y un sesgo asociado a esto. Es por eso que, en términos generales, un mayor tiempo de equilibrado puede generar resultados menos sesgados.
En cuanto a los estudios de eficiencia de derivatización, se observó que la señal del ion 256 m/z presentó un pico con tailing excesivo en comparación con el ion 329 m/z, coherente con la presencia de un grupo amino. La señal correspondiente al producto 1-TMS-creatinina no se detectó, posiblemente porque este producto no es lo suficientemente volátil para ingresar al sistema cromatográfico.
La eficiencia de derivatización también se estudió para el estándar interno, creatinina-d3, en este caso monitoreando los iones 332 m/z y 259 m/z. La mayor eficiencia de derivatización se alcanzó a mayores TD, en E6, E7, E8 y E9. Por otro lado, td parece no afectar significativamente la eficiencia de derivatización.
Las eficiencias de derivatización de creatinina y creatinina-d3 no fueron iguales en todos los casos; se encontró una eficiencia de derivatización entre 8 y 46% mayor para la creatinina. Esto presenta un problema, ya que la técnica de dilución isotópica por espectrometría de masas sostiene su alto desempeño analítico en que el comportamiento químico del analito (creatinina) y del estándar interno (creatinina-d3) es prácticamente igual. Se sospecha que la presencia de deuterio en el estándar interno desfavorece la reacción bajo algunas condiciones, por lo tanto, la eficiencia de derivatización disminuye. En vista de que la eficiencia de derivatización para creatinina es mayor que creatinina-d3, se buscaron las condiciones de relación de ED donde la relación fuera menor y se acercara a la unidad. Esto sucede cuando td disminuyó (E1, E2, E6 y E7), y en menor medida cuando tI aumentó (E2, E4, E7 y E9). El efecto favorable de la disminución de td en cuanto a la disminución de la relación ED y por ende del sesgo podría dar indicios de cinéticas de derivatización diferentes para creatinina y creatinina-d3. En este caso, es posible que la formación de 3-TMS-creatinina sea más rápida que la formación de 3-TMS-creatinina-d3 y a mayor tiempo de derivatización, mayor fue la diferencia entre el rendimiento de estos productos, induciendo un sesgo positivo. No obstante, fue posible encontrar condiciones donde la relación de ED permite un sesgo aceptable (E1, E4 y E6).
En la Tabla 5 se muestran los experimentos donde se optimizaron los factores para las diferentes variables de respuesta y la sumatoria de puntuación. Para los experimentos E2, E4, E7 y E9 se cumplieron dos condiciones de optimización para alguna de las respuestas, es decir, dos factores o combinación de factores tuvieron un efecto deseado sobre esta respuesta. Por este motivo, reciben doble puntuación en las respuestas que aplican.
Tabla 5. Resumen de experimentos de condiciones optimizadas para las diferentes variables de respuesta y su sumatoria.
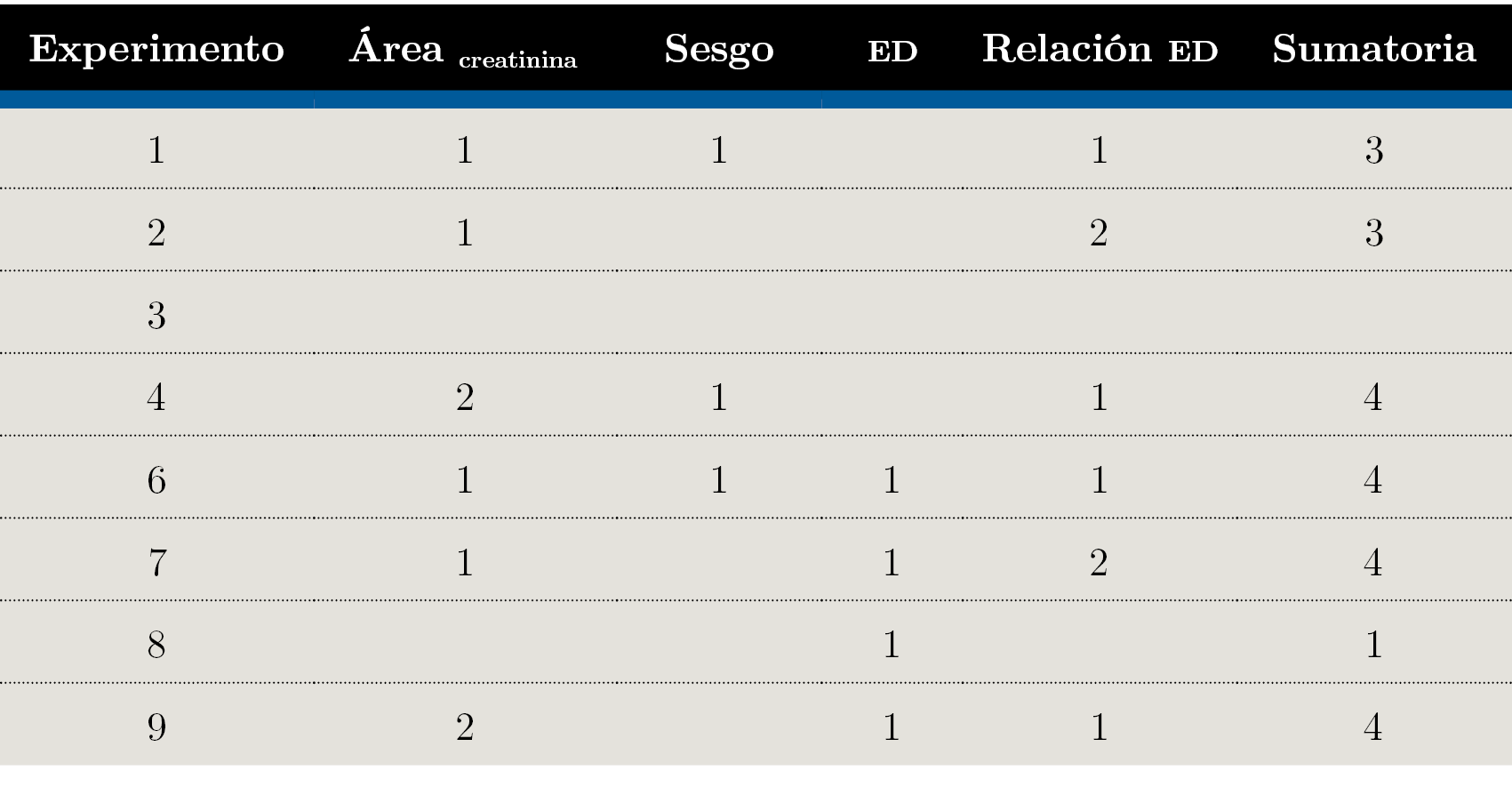
A partir de la tabla se observa que los experimentos que sumaron mayor puntaje en cuanto a criterios cumplidos son E4, E6, E7 y E9. No obstante, E7 y E9 no cuentan con las condiciones necesarias para la disminución del sesgo, un parámetro de mayor importancia porque afecta directamente la veracidad del método. Por esta razón se descartaron. El experimento E6 presentó como ventaja sobre E4 que la eficiencia de derivatización fue mayor y, adicionalmente, que el tiempo de análisis fue menor (E6: tI + td = 50 min vs 160 min para E4). Por lo tanto, se consideró que E6 presentó las mejores condiciones para este análisis en cuanto a veracidad y tiempo de análisis.
Conclusiones
Se evaluó el efecto de factores asociados a la etapa de equilibrado isotópico y derivatización sobre el método adaptado y se pudo encontrar condiciones óptimas para que los resultados presenten un sesgo adecuado al propósito (recuperación = 101%). El método adaptado tiene una mayor exactitud al implicar la preparación gravimétrica y mayor simplicidad, ya que permite el uso de un estándar interno disponible comercialmente, sin necesidad de sintetizarlo. Como ventaja adicional, este método propuesto reduce el tiempo de preparación de muestra en las etapas estudiadas a la mitad comparándolo al método de referencia (105 minutos versus 50 minutos). Una vez validado, este método adaptado podrá utilizarse para la asignación de valores de referencia trazables al sistema internacional de unidades a programas de evaluación externa de calidad y a materiales de referencia certificados. Estas herramientas de aseguramiento de la calidad serán de utilidad para los laboratorios clínicos nacionales que realizan mediciones empleando métodos de rutina.
Reconocimientos
Al equipo del Departamento de Metrología Química del Laboratorio Tecnológico del Uruguay.